Malaltia: SARS-CoV-2 / COVID-19
Col·laboració amb la Prof. Anna Planas Obrador de l'Institut d'Investigacións Biomèdiques de Barcelona (CSIC), el Dr. Israel Fernández Cadenas, de l'Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau, també de Barcelona, Marta López de Diego, del Centre Nacional de Biotecnologia (CSIC ), Fuencisla Matesanz, de l'Institut de Parasitologia i Biomedicina "López-Neyra", Lara Lloret de l'Institut de Física de Cantàbria (CSIC/Univ de Cantàbria) i Ignacio López Cabido del Centre de Supercomputació de Galícia (Xunta/CSIC).
Introducció
(tornar al començament)
Malaltia inicial: OPA10 (OMIM: #616732)
Col·laboració amb el Dr. Àngel Pérez Sempere, Neurologia. Hospital General d'Alacant.
Característiques genètiques: Forma familiar amb herència dubtosa però aparentment mendeliana recessiva. A la figura 1 es mostra l'arbre genealògic.
Introducció
(tornar al començament)
En aquest projecte estudiem el cas de dos germans, varons, actualment en edat adulta. Els dos germans (els dos únics fills d'aquesta família) presenten un quadre neurològic reportat des el primer any de vida, caracteritzat per una simptomatologia complexa. Aquests símptomes són iguals en tots dos pacients, amb algunes particularitats d'afectació. Els progenitors són sans i no mostren cap dels símptomes d'aquesta patologia (vegeu Figura 1). No s'han reportat més casos en antecessors dels afectats.
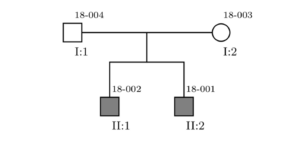
Figura 1
Arbre genealògic de la familia afectada per la malaltia.
El quadre clínic i, en conseqüència, el procés neurològic d'aquests pacients pot resumir-se en els següents símptomes:
• Retard en la capacitat motora apreciat des del primer any de vida. Els dos germans van començar a caminar a edat tardana, al voltant dels 3 anys.
• Atàxia progressiva. L'atàxia pot definir-se com una alteració de la coordinació de el moviment muscular voluntari. Com a tal, és un signe físic i per classificar-se dins d'una malaltia ha de investigar la seva etiologia. Generalment està causada per disfunció cerebel·losa o una alteració de l'impuls nerviós aferent a l'cerebel (Ashizawa i Xia, 2016). No obstant això, moltes atàxies no tenen diagnòstic, especialment aquelles amb una manifestació crònica i progressiva (com és aquest cas). Com a conseqüència, el germà més afectat va deixar de caminar en l'adolescència, mentre que l'altre és encara capaç de fer-ho, tot i que amb grans dificultats.
• Sensibilitat posicional disminuïda de manera severa en membres inferiors i lleument en membres superiors. Existeix en tots dos casos una dismetria severa a la prova dit-nas.
• Activitat cerebral reduïda: encefalograma lent.
• Reflexos abolits.
• Problemes visuals severs des de la infància amb atròfia òptica bilateral: el germà més afectat presenta ceguesa total mentre que el segon té visió molt reduïda. Aquesta condició s'ha reportat com la més cridanera i característica del procés neurològic, ja que les patologies relacionades amb aquest signe solen incloure molts la resta de símptomes.
• Parèsia de la mirada conjugada, bilateral: incapacitat parcial per moure tots dos ulls en diferents direccions.
• Disàrtria en ambdós casos, severa en el germà més afectat: dificultat en la programació motora de la parla i l'articulació de fonemes.
• Epilèpsia, controlada amb medicació. Les seves causes poden ser molt variades i heterogènies i moltes vegades es deu a la interacció entre diversos gens i factors ambientals, encara que també es presenta com a símptoma en determinats síndromes genètics.
• Retràs mental, amb dificultats de valoració icaracterització adequada degut als importants problemes visuals que tots dos malalts presenten. Es detecta un lleu deteriorament cognitiu.
Els estudis d'imatge (ressonància magnètica) donen resultats normals i sense patologies observables. El parènquima cerebel·lós i el tronc cerebral dels
pacients presenten morfologia normal, els ventricles cerebrals són simètrics i quart ventricle està centrat. Les intensitats de senyal són normals.També és destacable la inexistència d'afectació extraneurológica: S'han realitzat ecografies cardíaques i abdominals, proves d'exploració de fetge i
melsa i analítiques generals, resultant normals. Després de l'estudi de biòpsies musculars, també s'ha conclòs que no hi ha afectació muscular.
En la infància, els dos germans van ser diagnosticats d'Atàxia de Friedreich (FRDA). La FRDA és una malaltia neurodegenerativa d'herència autosòmica recessiva, el que podria ajustar-se a l'herència de
aquest cas. La causa genètica més comú és una expansió de la repetició d'un trinucleòtid GAA a l'intró 1 de el gen FXN (frataxina) (FRDA1) [OMIM #229300]. No obstant això, també hi ha una altra forma menys comuna on el gen responsable és el locus FRDA2 [OMIM %601992]. Es tracta d'una malaltia rara i amb
prevalença molt variable, poc comú en poblacions occidentals (1:20000 - 1:750000) i fins i tot menys en africanes i orientals (Burk, 2017).
La simptomatologia de la FRDA coincideix en diversos punts amb el quadre neurològic que es presenta en aquests germans, per la qual cosa el seu diagnòstic pot resultar acceptable en una època en què el diagnòstic genètic no estava tan implantat (tots dos van ser diagnosticats sense l'estudi genètic corresponent).
Les coincidències simptomatològiques són diverses: atàxia, disàrtria, atròfia òptica, debilitat en cames o sensibilitat posicional disminuïda. No obstant això, les proves genètiques realitzades més recentment han descartat la FRDA com a possible causa, així com altres atàxies cerebel·loses hereditàries.
Atròfia Òptica
(tornar al començament)
Les atròfies òptiques són un grup de neuropaties molt heterogènies. Es caracteritzen clínicament per una degeneració primària de les cèl·lules ganglionars de la superfície interna de la retina (RGCs), un grup de
neurones amb axó mielinitzat i els axons de les quals s'uneixen per formar el nervi òptic. La degeneració d'aquestes neurones desencadena l'atròfia de l'nervi òptic, donant lloc a les manifestacions clíniques de la neuropatia: disminució de l'agudesa visual, anomalies en el camp visual central i defectes en la percepció de colors, algunes formes poden arribar a la ceguesa (Orssaud, 2003; Gandhi, 2019). El dany al nervi òptic resulta irreversible.
Entre els canvis histopatològics més característics es troben la contracció o pèrdua dels axons i la beina de mielina de les RGCs, gliosi (proliferació de cèl·lules glials en lesions de sistema nerviós central) i una excavació fisiològica (depressió de l'ull) profunda, causant una exposició de la làmina
cribosa (estructura laminar que ocupa l'orifici posterior de l'escleròtica pel qual surt el nervi òptic) i ampliació de l'septe pial de l'ull (Gandhi, 2019). A això se li afegeix una ampliació de l'espai
subaracnoïdal (espai anatòmic pel qual circula el líquid cefaloraquidi).
No obstant això, l'OPA apareix en nombrosos casos associada a altres símptomes també de caràcter neurològic. En general, es classifica en tres grups: patològica, oftalmoscópica i etiològica. Al nostre cas, l'OPA hereditària (subclasse dins de l'etiològica) és la que presenta més interès. Esta subclasse presenta, al seu torn, diverses patologies i és habitualment sindròmica, amb símptomes extraoculars (Maresca et al., 2013).
Tot i que la majoria de vegades la patologia està causada per mutacions en el gen OPA1 (Gerber et al., 2017), hi ha molts altres gens descrits relacionats amb la malaltia (vegeu Taula 1). Dins de les atròfies òptiques s'inclouen les infantils o congènites (dominants o recessives), l'atròfia òptica hereditària de Behr [OMIM #210000], d'herència autosòmica recessiva, causada per mutacions homozigotes o heterozigòtiques compostes al gen OPA1, i l'atròfia òptica de Leber (LHON) [OMIM #535000], associada a mutacions en gens de l'ADN mitocondrial. En conseqüència, l'atròfia òptica és una neuropatia altament heterogènia. A la Taula 1 es resumeixen les diferents subformas, al costat de el gen afectat, localització,
herència i altres detalls.
Taula 1. Resum dels loci relacionats amb atròfies òptiques hereditàries.
| Localització | Fenotip | Herència | OMIM | Gen/Locus |
|---|---|---|---|---|
| 3q29 | Atròfia òptica 1 | AD | 165500 | OPA1 |
| Xp11.4-p11.21 | Atròfia òptica 2. Lligada a l'X | XL | 311050 | OPA2 |
| 19q13.32 | Atròfia òptica 3 amb cataractes | AD | 165300 | OPA3 |
| 18q12.2-q12.3 | Atròfia òptica 4 | - | 605293 | OPA4 |
| 12p11.21 | Atròfia òptica 5 | AD | 610708 | DNM1L |
| 8q21-q22 | Atròfia òptica 6 | AR | 258500 | OPA6 |
| 11q14.1 | Atròfia òptica 7 | AR | 612989 | TMEM126A |
| 16q21-q22 | Atròfia òptica 8 | AD | 616648 | OPA8 |
| 22q13.2 | Atròfia òptica 9 | AR | 616289 | ACO2 |
| 6q11 | Atròfia òptica 10 amb o sense atàxia, retràs mental i convulsions | AR | 616732 | RTN4IP1 |
| 10p12.1 | Atròfia òptica 11 | AR | 617302 | YME1L1 |
Malgrat la seva heterogeneïtat genètica, podriem dir que la major part de les formes d'atròfia òptica hereditàries compartixen vies moleculars comunes: estan causades per defectes en proteïnes amb funcions mitocondrials. En molts dels casos, hi ha alteracions en subunitats dels complexos de la cadena de transport
electrònic mitocondrial, com és el cas de l'atròfia de Leber. En altres casos, les proteïnes afectades intervenen en la dinàmica mitocondrial i en mecanismes de fusió i fissió de les membranes externa i interna del mitocondri, com és el cas d'OPA1 o de DNM1L (OPA5), GTPases relacionades amb
dinaminas (Gerber et al., 2017). També és notori el cas d'OPA9, causada per mutacions en el gen
ACO2 (aconitasa 2), que catalitza la interconversió de citrat a isocitrat en el segon pas de l'Cicle de Krebs, donant lloc a atròfia òptica amb encefalopatia i atròfia cerebelar (Metodiev et al., 2014).
En conclusió, es pot dir que hi ha cinc causes moleculars descrites (tot i que encara hi ha diverses desconegudes) d'atròfia òptica: dèficit en el complex I mitocondrial, producció excessiva d'espècies reactives d'oxigen (ROS), fallades en la dinàmica mitocondrial, apoptosi mitocondrial, dèficit de la producció d'ATP i biogènesi mitocondrial (Maresca et al., 2013), moltes d'elles interconnectades i relacionades.
No obstant això, molts dels gens mostrats a la Taula 1 no tenen una funció coneguda, malgrat el seu paper en aquestes malalties. Per exemple, TMEM126A és una proteïna transmembrana localitzada a la membrana interna mitocondrial, d'implicació en OPA7 però de funció incerta
(Meyer et al., 2010). Existeixen diferents variants descrites en aquest gen donant lloc a un fenotip OPA, algunes de recent descobriment (Kloth et al., 2019). Fins i tot es desconeix quin és el gen responsable d'alguns d'ells, com OPA2 o OPA4 entre d'altres.
En conclusió i, malgrat el seu major o menor coneixement, les proteïnes anteriorment esmentades es troben altament expressades o formen part del mitocondri. Aquest fet podria oferir suport a la hipòtesi que l'atròfia òptica, com molts altres dels símptomes amb els quals sol presentar-se, pot ser
una marca subjacent d'un defecte en la dinàmica, síntesi energètica i funcionament mitocondrial. Malgrat tot, és important assenyalar que també existeixen gens d'expressió no mitocondrial amb certa rellevància en atròfia òptica, encara que en molta menor proporció.
Aproximació experimental
(tornar al començament)
Per identificar el gen responsable de l'aparició de la malaltia en esta família, anem a començar per l'anàlisi d'exoma complet.
La seqüenciació de exomes s'ha convertit, des de fa una dècada (i cada vegada més gràcies a la reducció del cost de NGS) en una poderosa eina cost-efectiva per disseccionar les causes genètiques de Hi ha molts casos on s'empren aproximacions, tant experimentals com analítiques, basades en aquesta estratègia.
Actualment, WES s'empra principalment per detectar variants rares en pacients amb sospites de patir qualsevol tipus de trastorn genètic mendelià i quan una aproximació basada en múltiples gens és laboriós, costós o simplement no és possible al desconèixer qualsevol relació genotip-fenotip possible. Per a aquest grup de trastorns, WES ha demostrat ser eficient, oferint un diagnòstic per a una quantitat significativa de pacients (Brown i Meloche, 2016). Especial menció mereix
el cas descrit a Science per Kaiser (2010), per ser un dels primers exemples, al costat de el ja comentat abans, i per determinades similituds amb el fenotip buscat en aquest cas.
També és destacable com les causes genètiques d'altres malalties similars, subtipus d'OPA, han estat identificades gràcies a WES (Metodiev et al., 2014; Angebault et al., 2015; Hartmann et al., 2016; Gerber et al., 2017). El WES no només ens permet buscar noves variants i gens relacionats, sinó que al mateix temps també ens permet avaluar altres gens potencialment relacionats, ja descrits anteriorment com OPA.
L'ús de mètodes tradicionals, com la seqüenciació Sanger, obliga a conèixer a priori tots els gens per analitzar (Harding i Robertson, 2014). Per això, WES suposa un clar avantatge com a eina de diagnòstic, ja que no es necessita una pre-selecció de gens i permet una anàlisi completa de diversos gens que en principi poden semblar no associats a la malaltia.
Projectes de seqüenciació de exomes a gran escala (n> 500) han reportat rendiments de l'25-30% en la recerca del genotip causant de la malaltia (Farwell et al., 2015). No obstant això, assajos més
recents han augmentat el rendiment fins al 43% (Baldridge et al., 2017) i fins i tot fins al 57% quan
s'empra en grups seleccionats de pacients amb malalties monogèniques sospitades (Mak et al., 2018).
Anteriorment, els estudis de lligament eren la principal eina per a l'estudi de malalties rares, però, trastorns molt rars o heterogenis i amb famílies petites no eren adequats per aquesta estratègia. WES no compta amb aquesta limitació, i fins i tot pot detectar variants associades a malalties que no havien estat detectades a estudis anteriors per la seva sensibilitat (Tétreault et al., 2015).
D'altra banda, també s'ha mostrat la superioritat de WGS / WES enfront d'altres tècniques com els xips (Arrays, micromatrius) cromosòmics (CMA, que inclouen xips d'hibridació genòmica comparada i xips de SNPs). Una
metanàlisi recent portat a terme per Clark i col·laboradors (2018) mostra com WGS / WES són
quantitativament millors que CMA (P <0,0001) per al diagnòstic de nens amb sospites de malalties genètiques. En aquest sentit, seria convenient considerar WGS / WES com a prova genòmica de primera línia, en lloc dels CMA, més establerts fins ara. Altres tècniques emprades per al diagnòstic genètic són els panells de gens o l'anàlisi de variants en el nombre de còpies, encara que són de major utilitat quan a priori ja se sospita la relació de certs gens i la malaltia és més coneguda.
Resultats
(tornar al començament)
En procés
Text modificat del TFG de Gonzalo Mercado Vico per al curs 2018/2019.